Kategóriák
- Vizsgálati kisokos (5)
- Egyéb kategória (18)
- Hírek (115)
- Betegségek (54)
TARDBP és FUS génmutáció specifikus terápiás fejlesztések ALS-ban
Az amiotrófiás laterálszklerózis, röviden ALS egy izomsorvadással járó neurodegeneratív megbetegedés. Ismert, hogy az ALS esetek mintegy 10 százaléka mutat családi halmozódást, az esetek további 90 százaléka sporadikus eredetű. Habár a leggyakoribb génmutációk az európai ALS betegek körében a SOD1 és a C9orf72 génben fordulnak elő, nem elhanyagolható más gének és ezek mutációinak szerepe.
A TARDBP gén által kódolt fehérjét TDP-43-nak nevezzük (43 kilodalton molekulasúlyáról kapta a nevét). A TARDBP génmutáció a családi halmozódású ALS esetek mintegy 4%-ában, míg a sporadikus esetek mintegy 1.5%-ában detektálható. Egyes adatok szerint a TARDBP mutációk leggyakrabban dél-európai ALS betegek körében fordulnak elő. A fehérje sok ALS-hoz kapcsolt sejten belüli útvonal működésében lát el fontos szerepet, pl. részt vesz RNS-kötő fehérjeként az RNS-ek metabolizmusában, részt vesz a sejten belüli fehérje képződési és lebomlási egyensúly létrehozásában és ennek fenntartásában. A TDP-43 fehérje diszfunkciója vezethet mitokondriális működészavarokhoz és a DNS hibajavítási folyamatainak zavarához.
Élettani körülmények között a TDP-43 fehérje a sejtek magjában található, ott látja el a feladatát. Kóros körülmények között a TDP-43 fehérje kicsapódik, és az idegsejten belül zárványtestet képez, ami meggátolja a sejtet az élettani funkciójának ellátásában és ezáltal a sejt elhal.
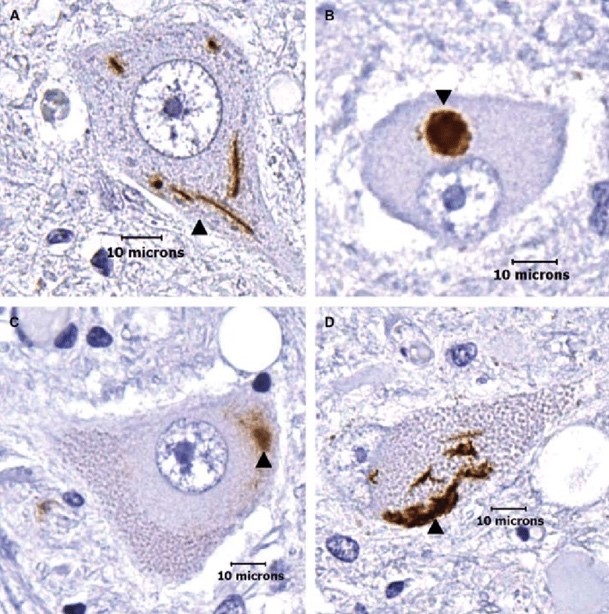
A FUS gén egy fused-in-sarcoma nevű fehérjét kódol, aminek neve arra utal, hogy először egy bizonyos kötőszöveti daganattípusban (a sarcomaban) azonosították a fehérjét. A fehérje egy RNS-kötő molekula, ami szintén részt vesz a DNS hibajavító folyamataiban és a fehérje egyensúly sejten belüli fenntartásában. Emellett fontos szerepet tölt be az RNS-ek érési folyamataiban, és az RNS-ek transzportfolyamataiban. A fehérje túlzott képződése a TDP-43 fehérjéhez hasonlóan sejten belüli zárványok képződéséhez és ezáltal sejthalálhoz vezet. A FUS gén variánsai ritkábban fordulnak elő európai származású ALS betegek körében, a familiáris esetek mintegy 4%-ában, míg a sporadikus esetek kevesebb, mint 1%-ában fordulnak elő a gén mutációi. A FUS génmutációt hordozó betegek földrajzi eloszlása kevésbé homogén, egyes népcsoportokban akár 10% is lehet a FUS mutációt hordozó ALS betegek aránya.
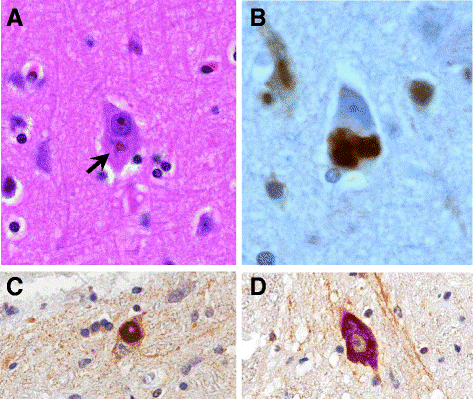
Jelenleg több terápiás lehetőség is fejlesztés alatt áll a TARDBP illetve a FUS mutációval élő ALS betegek számára.
A TARDBP mutációt hordozó betegek számára fejleszti a Dewpoint Therapeutics cég a „condensate modifying compound” (aggregátum módosító vegyület) néven említett molekuláját, ami a fehérjeaggregátumok lebontását célozza meg. A fejlesztés jelenleg még csak a preklinikai fázisban tart, és a Target ALS alapítvány támogatását élvezi. A cég közlése szerint a molekula effektíven bontja le a sejtplazmában felhalmozódott fehérjét és visszajuttatja a TDP-43-at a sejtmagba és visszaállítja a fehérje eredeti funkcióját. A molekulát jelenleg még csak betegeredetű, laborban növesztett mozgató idegsejteken, valamint egérmodellen tesztelik. Egérmodell vizsgálatokkal is biztató eredmények születtek: csökkentette a kísérleti szer a neurodegeneráció és az idegrendszeri gyulladás markereinek szintjét, valamint csökkent a TDP-43 aggregátumok mennyisége is a kísérleti állatokban.
Kölnben dolgozó kutatók azt találták, hogy két fehérje, a H1.2 és a PARP1 fehérjék, amikor interakcióba lépnek, fokozódik a FUS tartalmú aggregátumok képzése. Ezzel ellentétben viszont, amikor gilisztamodellben csökkentették a H1.2 és a PARP1 fehérjék kifejeződését, csökkent a FUS tartalmú aggregátumok képződésének üteme. Ez arra enged következtetni, hogy a FUS-a H1.2- a PARP1 fehérjék egy funkcionális hálózatot alkotnak és ennek a fehérje hálózatnak a befolyásolása hatással lehet a FUS aggregátumok formálódásának ütemére. A kutatók reményüket fejezték ki, hogy a felfedezésüket később a klinikumban is hasznosítani fogják.
Források:
- Bodansky, Aaron et al. “TDP-43 and ubiquitinated cytoplasmic aggregates in sporadic ALS are low frequency and widely distributed in the lower motor neuron columns independent of disease spread.” Amyotrophic lateral sclerosis : official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases vol. 11,3 (2010): 321-7. doi:10.3109/17482961003602363
- Nolan, Matthew et al. “Pathogenesis of FUS-associated ALS and FTD: insights from rodent models.” Acta neuropathologica communications vol. 4,1 99. 6 Sep. 2016, doi:10.1186/s40478-016-0358-8
- Mackenzie, Ian Ra et al. “TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia.” The Lancet. Neurology vol. 9,10 (2010): 995-1007. doi:10.1016/S1474-4422(10)70195-2
- https://dewpointx.com/tdp-43-condensate-modulation-rescues-tdp-43-loss-of-function-in-als-patient-derived-motor-neurons-and-mouse-models-of-tdp-43-proteinopathy/
- Alirzayeva, Hafiza et al. “ALS-FUS mutations cause abnormal PARylation and histone H1.2 interaction, leading to pathological changes.” Cell reports vol. 43,8 (2024): 114626. doi:10.1016/j.celrep.2024.114626
Szerkesztette: dr. Nagy Zsófia Flóra
2024. október 07.