Kategóriák
- 1. Izomdisztrófiák (47)
- 2. Miopátiák (4)
- 3. Ioncsatorna betegségek (1)
- 4. Mozgatóideg betegségek (36)
- 5. Metabolikus miopátiák (1)
- 6. Perifériás idegbántalmak (5)
- 7. Gyulladásos miopátiák (1)
- 8. Izom-ideg átkapcsolódási zavarok (4)
- 9. Endokrin miopátiák (0)
- 10. Mitokondriális miopátiák (0)
- 11. Diagnosztika (8)
- 12. Gyógyszeres terápia (35)
- 13. Dietetika (3)
- 14. Ortopédia (1)
- 15. Pszichológia (3)
- 16. Légzésterápia (1)
- 17. Klinikai vizsgálatok (36)
- 18. Gyógytorna (4)
- 19. Szociális ellátások (1)
- 20. Egyéb (23)
Az amiotrófiás laterálszklerózis (ALS) genetikai háttere
Vérrokonok hasonló amiotrófiás laterálszklerózis-szerű tünetei hívták fel a figyelmet az ALS lehetséges genetikai hátterére. Családi halmozódásúnak tekinthető az ALS (familiáris ALS, fALS), amennyiben legalább egy elsőfokú vagy másodfokú rokont korábban ALS-sal diagnosztizáltak. Az fALS esetek az összes ALS eset mintegy 10%-át teszik, ki a további kb 90% sporadikusnak tartható (sALS).
Ismertek testi- és ivari kromoszómához kötötten öröklődő ALS formák, valamint domináns módon (egy hibás génváltozat okozza) illetve recesszíven (két hibás génváltozat kell a betegség megjelenéséhez) öröklődő géneltérések is. Változó ezen géneltérések penetranciája is: ismertek teljes penetranciát mutató eltérések (tehát, amennyiben a géneltérés jelen van, biztos, hogy a betegség is ki fog alakulni) illetve csökkent penetranciájú génmutációk is (a mutáció hordozása esetén sem 100% az esélye, hogy a tünetegyüttes ki fog alakulni az alany élete során). Csupán a pontos géneltérés ismerete mellet tudunk ismétlődési kockázatot becsülni az utódgeneráció tekintetében.
Az amiotrófiás laterálszklerózis, röviden ALS, hátterében az első gént 1993-ban azonosították, ez volt a SOD1 (szuperoxid dizmutáz). A gén egy antioxidáns szerepet jásztó enzimet kódol, ami a sejteket védi a szabadgyökök roncsoló hatásától. A SOD1 mutációk következtében túl stabil fehérjetermék jön létre, amit nem tudnak lebontani az erre hivatott sejtszervecskék, ezért ez a fehérje felhalmozódik és kicsapódik, ami a sejt halálához vezet.
2008-ban két független kutatócsoport nagyjából egy időben írta le a C9orf72 gén hat egységből álló ismétlődéshossz változását (repeat expanzió). Ez a genetikai eltérés az európai populációban a sporadikus ALS esetek mintegy 7%-áért tehető felelőssé, míg a családi halmozódást mutató esetek akár 67%-ában is azonosítják a C9orf72 gén hibáját. A gén által kódolt fehérje szerepe mindezidáig nem teljesen megértett, feltételezhetően a sejten belüli RNS transzport folyamatokban játszik szerepet. Az egészséges emberek a C9orf72 gén ezen szakaszán maximum 20 darab, hat egységből álló nukleotidismétlődést hordoznak. Ez ALS betegekben akár 1000 egység is lehet. Ez a meghosszabbodott génszakasz a DNS-ben tárolt információ fehérjére való lefordítása közben hajlamos önmagához tapadni és hajtűszerű, összegabalyodott struktúrákat képezni. Ezek a képződött fehérjék is ellenállóak a lebontó folyamatokkal szemben, így felhalmozódnak, és jelenlétükkel túlterhelik a sejtet és akadályozzák a sejt normális működését.
További, jelentős ALS-sal kapcsolatba hozott gének a következők: TARDBP, FUS, TBK1, NEK1.
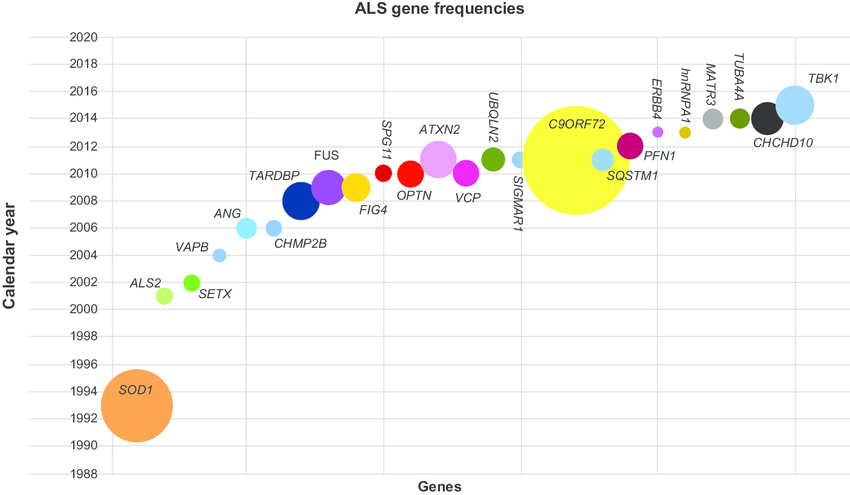
Az ALS-sal kapcsolatba hozott gének csoportosítása a betegség és a gén kapcsolatán alapul. Eddig több mint 30 kóroki ALS gént írtak le, melyek kapcsolata a betegség kialakulásával jól megalapozott. Több független kutatás is kapcsolatba hozta variánsaikat az ALS-sal őket, valamint nagyrészt ismert, hogy milyen módon kapcsolódnak az ALS összetett pathomechanizmusához, milyen folyamat módosításával járulnak hozzá a betegség kialakulásához. Az ALS kandidáns gének kapcsolata a betegséggel még nem teljesen tisztázott. Több mint 120 gén tartozik ebbe a csoportba, ám számuk köszönhetően az ALS genetikai hátterének felderítésére irányuló fáradhatatlan kutatásoknak, egyre nő. A legaktuálisabb génlista az ALSoD adatbázisban fellelhető.
Az ALS genetikai hátterének vizsgálatát nehezíti, hogy a familiáris esetek körülbelül 60%-ában, míg a sporadikus esetek csupán 15%-ában azonosítható a kóroki genetikai eltérés, valamint nagy szerepe van a populációs és etnikai különbségeknek is. Ismertek olyan variánsok, amelyek egy populációban felettébb gyakoriak, míg egy másik populációban egyáltalán nem fordulnak elő (pl. egyes zárt közösségek, mint pl. Málta, Szardínia). Ezen felismerés miatt fontosak az ALS betegségben a populáció specifikus genetikai vizsgálatok.
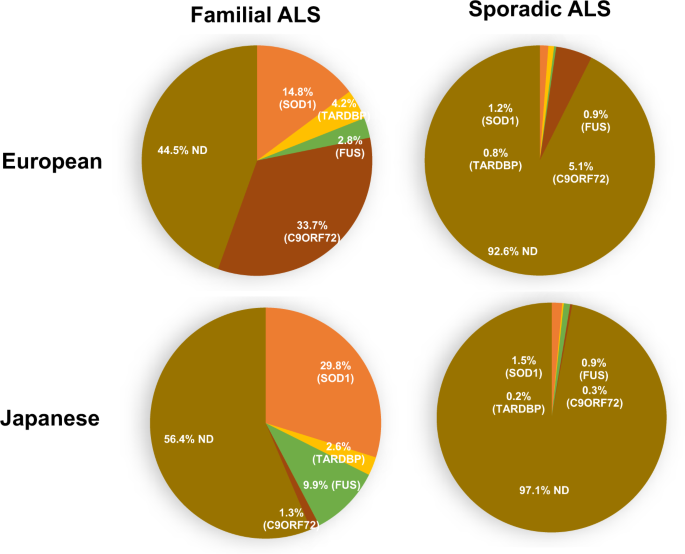
Korábban Magyarországon végzett ALS genetikai vizsgálatok
Mindezidáig 5 tanulmány jelent meg a nemzetközi szakirodalomban, ami a magyar ALS-ban szenvedő betegeket vizsgálta. Ezek között voltak célzott, egy-egy gént specifikusan vizsgáló tanulmányok is, valamint készült 107 magyar ALS beteg bevonásával egy nagyobb lefedőképességű panel- illetve teljes exom szekvenálásos vizsgálat is.
Kijelenthető, hogy a magyar, sporadikus megjelenésű ALS-sal diagnosztizált betegek körében a leggyakoribb genetikai eltérés a C9orf72 gén ismétlődéshossz változása. A vizsgált betegek kb. 9.5%-ánál igazolódott egy kórosan hosszú allél. Nagyobb betegszámú, Olaszországban végzett vizsgálatokban ez az arány 5% körül alakul, míg egy belga betegeket vizsgáló tanulmányban csupán a betegek 3.8%-a hordozta ezt az eltérést. Nem pontosan tudjuk jelenleg, hogy mi az oka, hogy Magyarországon gyakoribb a C9orf72 gén repeat expanziója, mint tőlünk nyugatabbra fekvő országokban.
A C9orf72 gén eltéréseit követik követi gyakoriságban a SOD1 gén mutációi. Ezek közül is a leggyakoribb a gén által kódolt fehérje 145. aminosavának cseréje, amit a szakirodalomban p.L145F kóddal illetnek. Magyarországon eddig 7 ALS betegben azonosították ezt a variánst. A mutációt egy magyar származású tudós által vezetett USA-beli kutatócsoport funkcionális vizsgálatnak is alávetette. Azt találták, hogy a mutáció következtében a gén által kódolt enzimtermészetű fehérje aktivitása csupán kismértékben csökken. Ez magyarázhatja a mutációt hordozó betegeknél tapasztaltakat: ezt a mutációt hordozó betegek idősebben veszik észre az első tüneteket, és az átlagnál lassabb lefolyású betegséget tapasztalnak. A p.L145F mutációval élő betegek esetében gyakrabban fordul elő kognitív hanyatlás is.
Genetikai tanácsadás
Az ALS diagnózisa esetén javasolható a genetikai tanácsadáson történő részvétel, ahol a klinikai genetikus kolléga részletesen tájékoztatja a beteget a jelenleg elérhető genetikai vizsgálatokról, ezek lehetséges kimeneteléről, a tudáshoz, illetve a nemtudáshoz való jogról. Ezen információk tudatában a beteg tájékozott döntést hozhat a genetikai vizsgálat elvégzéséről. SOD1 génmutáció által okozott ALS formára már hazánkban is rendelkezésre áll genetikai alapú terápia köszönhetően a korai hozzáférési programnak. Továbbá a C9orf72 génhiba által okozott ALS kezelésére fejlesztett genetikai alapú gyógyszer jelenleg fázis 2b állapotú klinikai vizsgálatban tart külföldön, az elkövetkező években várható a szer hazai megjelenése is klinikai vizsgálatok keretein belül.
Irodalom:
- http://www.alsod.ac.uk
- https://www.nature.com/articles/s10038-022-01055-8
- Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014 Jan;17(1):17-23. doi: 10.1038/nn.3584. Epub 2013 Dec 26. PMID: 24369373; PMCID: PMC4544832.
- Brenner D, Weishaupt JH. Update on amyotrophic lateral sclerosis genetics. Curr Opin Neurol. 2019 Oct;32(5):735-739. doi: 10.1097/WCO.0000000000000737. PMID: 31335339.
- Gal J, Kuang L, Barnett KR, Zhu BZ, Shissler SC, Korotkov KV, Hayward LJ, Kasarskis EJ, Zhu H. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 2016 Oct;132(4):563-76. doi: 10.1007/s00401-016-1601-x. Epub 2016 Aug 1. PMID: 27481264; PMCID: PMC5023729.
- Tripolszki K, Gampawar P, Schmidt H, Nagy ZF, Nagy D, Klivényi P, Engelhardt JI, Széll M. Comprehensive Genetic Analysis of a Hungarian Amyotrophic Lateral Sclerosis Cohort. Front Genet. 2019 Aug 16;10:732. doi: 10.3389/fgene.2019.00732. PMID: 31475037; PMCID: PMC6707335.
- Tripolszki K, Danis J, Padhi AK, Gomes J, Bozó R, Nagy ZF, Nagy D, Klivényi P, Engelhardt JI, Széll M. Angiogenin mutations in Hungarian patients with amyotrophic lateral sclerosis: Clinical, genetic, computational, and functional analyses. Brain Behav. 2019 Jun;9(6):e01293. doi: 10.1002/brb3.1293. Epub 2019 Apr 25. PMID: 31025543; PMCID: PMC6576160.
- Tripolszki K, Csányi B, Nagy D, Ratti A, Tiloca C, Silani V, Kereszty É, Török N, Vécsei L, Engelhardt JI, Klivényi P, Nagy N, Széll M. Genetic analysis of the SOD1 and C9ORF72 genes in Hungarian patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2017 May;53:195.e1-195.e5. doi: 10.1016/j.neurobiolaging.2017.01.016. Epub 2017 Jan 29. PMID: 28222900.
- Tripolszki K, Török D, Goudenège D, Farkas K, Sulák A, Török N, Engelhardt JI, Klivényi P, Procaccio V, Nagy N, Széll M. High-throughput sequencing revealed a novel SETX mutation in a Hungarian patient with amyotrophic lateral sclerosis. Brain Behav. 2017 Mar 15;7(4):e00669. doi: 10.1002/brb3.669. PMID: 28413711; PMCID: PMC5390843.
- Nagy ZF, Pál M, Salamon A, Kafui Esi Zodanu G, Füstös D, Klivényi P, Széll M. Re-analysis of the Hungarian amyotrophic lateral sclerosis population and evaluation of novel ALS genetic risk variants. Neurobiol Aging. 2022 Aug;116:1-11. doi: 10.1016/j.neurobiolaging.2022.04.002. Epub 2022 Apr 9. PMID: 35525134.
Készítette: dr. Nagy Zsófia Flóra
2024. március 04.